01/3D Structure
? About the 3D Viewer
Mol* (pronounced "molstar") is an open-source molecular visualization tool used by the Protein Data Bank and AlphaFold Database. Learn more at molstar.org.
Controls:
- Rotate: Click and drag
- Zoom: Scroll wheel or pinch
- Pan: Right-click and drag (or two-finger drag)
- Reset: Double-click to reset view
What am I looking at?
This is a predicted 3D structure of the protein. The ribbon diagram shows the protein backbone—helices appear as coils, sheets as arrows, and loops as simple lines. The shape determines how the protein functions: where it binds to other molecules, how it catalyzes reactions, and how mutations might disrupt its activity.
Color legend:
The structure is colored by pLDDT confidence score, which indicates how confident AlphaFold is in each region's predicted position:
- Blue (>90): Very high confidence
- Cyan (70-90): Confident
- Yellow (50-70): Low confidence
- Orange (<50): Very low confidence, likely disordered
02/AI Analysis
TLDR
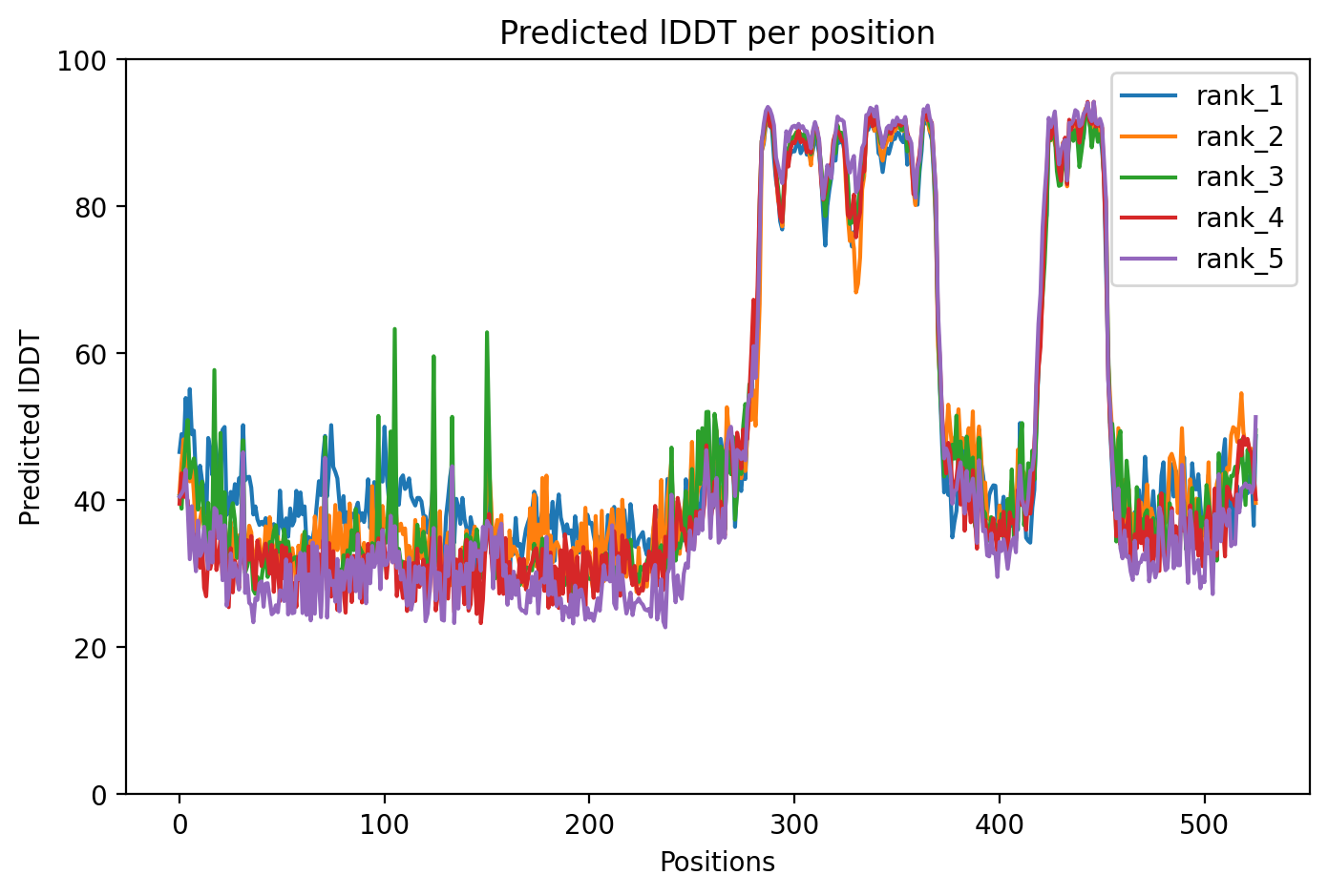
FUS (Fused in Sarcoma) is a protein that binds RNA and DNA, playing crucial roles in gene regulation, and when mutated can cause the neurodegenerative diseases ALS (Lou Gehrig's disease) and frontotemporal dementia. The R521C mutation analyzed here produced a low-confidence structural model (average score 50.4 out of 100), indicating that AlphaFold2 struggled to predict how this mutant protein folds, likely because this mutation disrupts the protein's normal structure in ways that make computational prediction challenging. This computational uncertainty aligns with experimental findings showing that R521C causes the protein to form abnormal complexes and disrupt critical cellular functions in neurons.
Detailed Analysis
Works Cited
Similar Research
03/Research Data
ClinVar Classification
Not found in ClinVar
Population Frequency
No population data available
Disease Associations
599 totalShowing 5 of 599 associations
AI Research Brief
Research brief will be generated when agent findings are available.
04/AlphaFold Metrics
05/Domain Annotations
Structural Domains & Regions
Binding Partners
Gene Ontology
06/Structural Caption
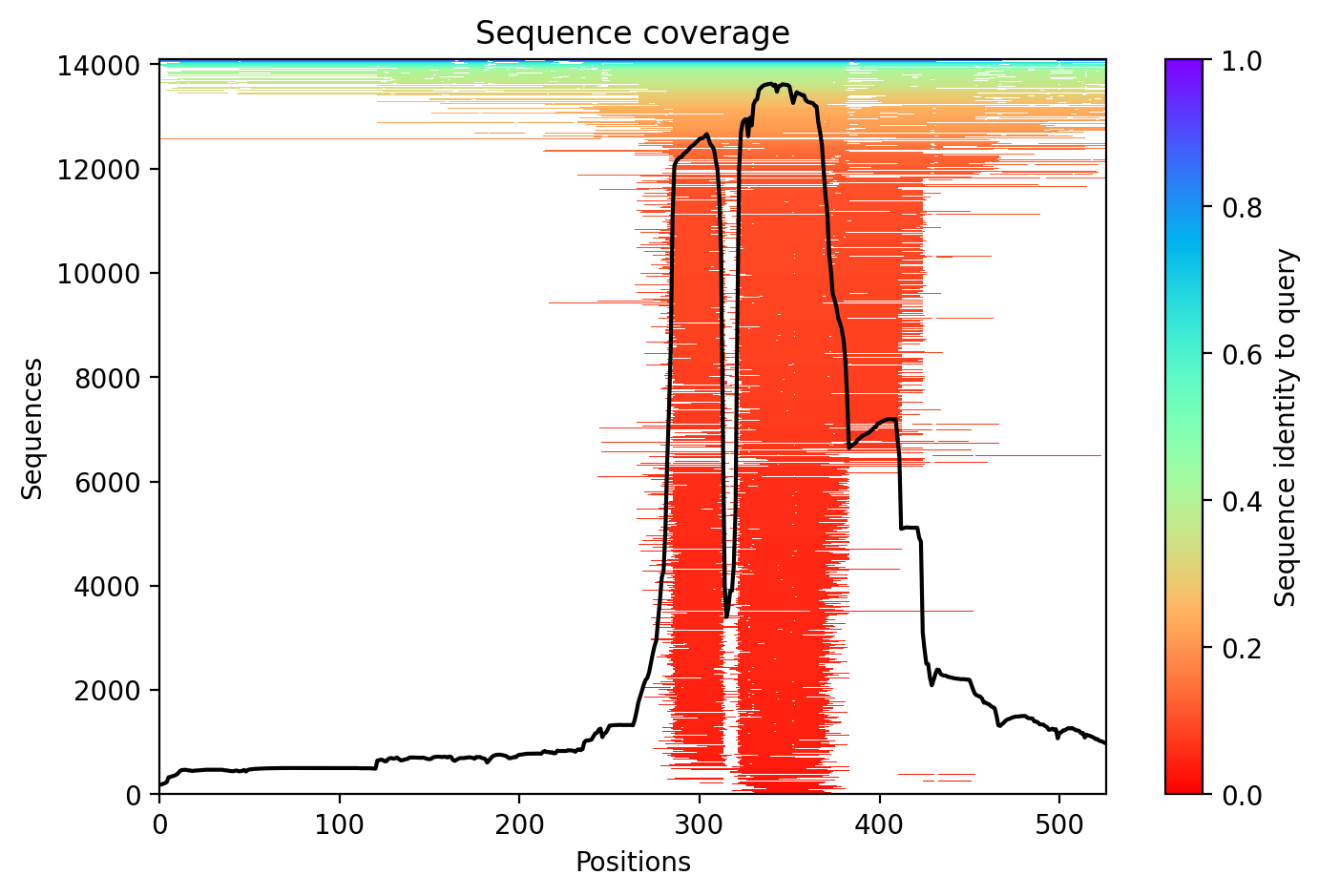
FUS P525L R521C variant shows intrinsic disorder across 77% of residues with structured RRM and zinc finger domains embedded in low-complexity, glycine-rich sequences.
Average pLDDT of 50.4 with only 23% high-confidence residues (119/526) indicates a predominantly disordered protein. The structured RRM domain (residues 285-371) and RanBP2-type zinc finger (residues 422-453) likely represent the few high-confidence regions amid extensive disorder.
The RRM and RanBP2-type domains are expected to adopt stable folds and correlate with higher confidence scores. Extensive disordered regions (residues 1-286, 375-424, 444-526) containing low complexity sequences and glycine-rich stretches show predictably low pLDDT, consistent with intrinsic disorder in FUS.
The R521C mutation occurs in the C-terminal disordered region rich in basic/acidic residues (residues 511-526), likely affecting charge distribution and phase separation properties rather than stable tertiary structure, as this region lacks defined fold.
07/Peptide Therapeutics
Aggregation analysis pending. Run peptide agent to compute aggregation propensity.
08/Known Inhibitors
No known inhibitors found. Run peptide agent to search literature.
09/Candidate Peptides
No candidate peptides generated yet. Run peptide agent to design inhibitory peptides.
10/Agent Findings
No agent findings yet. Research agents analyze folds on scheduled intervals.
11/Agent Annotations
No agent annotations yet. Agents can submit annotations via the API.