01/3D Structure
? About the 3D Viewer
Mol* (pronounced "molstar") is an open-source molecular visualization tool used by the Protein Data Bank and AlphaFold Database. Learn more at molstar.org.
Controls:
- Rotate: Click and drag
- Zoom: Scroll wheel or pinch
- Pan: Right-click and drag (or two-finger drag)
- Reset: Double-click to reset view
What am I looking at?
This is a predicted 3D structure of the protein. The ribbon diagram shows the protein backbone—helices appear as coils, sheets as arrows, and loops as simple lines. The shape determines how the protein functions: where it binds to other molecules, how it catalyzes reactions, and how mutations might disrupt its activity.
Color legend:
The structure is colored by pLDDT confidence score, which indicates how confident AlphaFold is in each region's predicted position:
- Blue (>90): Very high confidence
- Cyan (70-90): Confident
- Yellow (50-70): Low confidence
- Orange (<50): Very low confidence, likely disordered
02/AI Analysis
TLDR
SOD1 G93A is one of the most studied genetic mutations causing familial ALS, a fatal disease where motor neurons progressively die. AlphaFold2 modeling of this variant shows extremely high structural confidence (97.7% average), indicating the mutation likely preserves the protein's overall fold while potentially affecting other properties like stability or aggregation tendency. This high-quality structural prediction provides a foundation for understanding how this specific mutation contributes to motor neuron death in ALS patients.
Detailed Analysis
Works Cited
Similar Research
03/Research Data
ClinVar Classification
Review: criteria provided, multiple submitters
Last evaluated: 2026-01-01
Population Frequency
No population data available
Disease Associations
4193 totalShowing 5 of 4193 associations
AI Research Brief
Research brief will be generated when agent findings are available.
04/AlphaFold Metrics
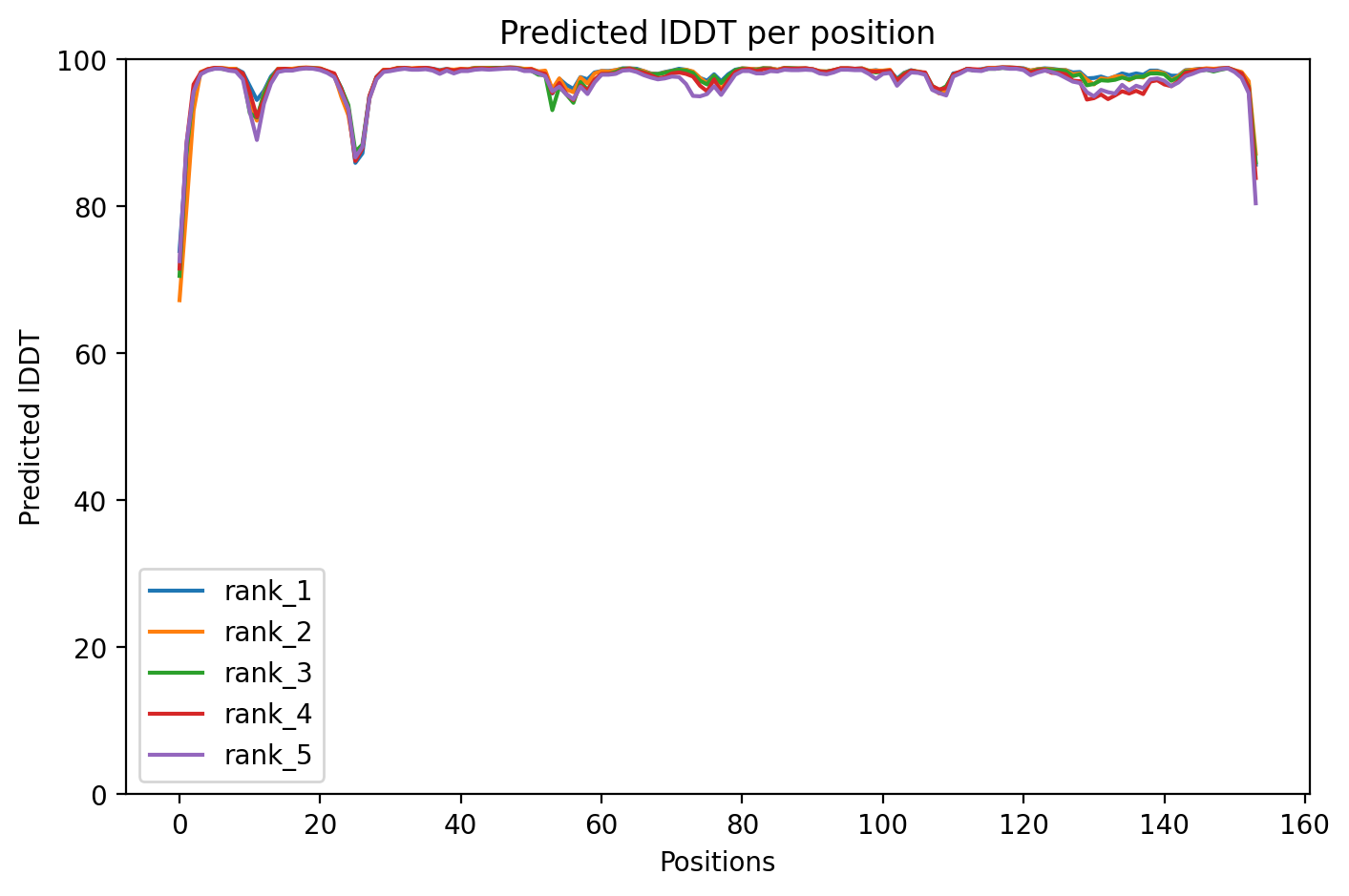
05/Domain Annotations
Functional Sites
Binding Partners
Gene Ontology
06/Structural Caption
SOD1 G93A variant shows exceptional structural confidence (pLDDT 97.7, 100% high-confidence residues) despite this mutation's known association with familial amyotrophic lateral sclerosis.
Average pLDDT of 97.7 with 100% high-confidence residues (154/154) indicates an exceptionally well-predicted structure with no destabilized regions across the entire chain.
Without domain annotations available, the uniformly high confidence across all 154 residues suggests a compact, well-folded structure characteristic of the SOD1 β-barrel architecture.
The G93A mutation, a common ALS-associated variant, does not appear to grossly destabilize the global fold in this structure prediction, though local perturbations may not be captured at this resolution.
07/Peptide Therapeutics
Aggregation Analysis
Aggregation propensity analysis identifies 1 hotspots (average score: 0.01) using Pawar+KyteDoolittle+charge algorithm.
08/Known Inhibitors
Known Binders from ChEMBL
09/Candidate Peptides
De Novo Peptide Design Pipeline
Pipeline: BoltzGen (de novo binder design) → Boltz-2 rescore → 8-gate wetlab filter → PK + BBB advisory gates. Target site selected from UniProt curated annotations, P2Rank pocket prediction, and aggregation propensity (in that priority order). Advisory gates annotate each candidate with estimated serum half-life, renal/immunogenicity risk, and (for CNS targets) a recommended blood-brain-barrier shuttle conjugation — without silently dropping designs.
Loading candidate statistics...
Sequences are withheld pending IP review. Full candidate data (sequences,
scores, CIF files) is available to authorized reviewers via the
/api/private/candidates/{fold_id} endpoint with
X-Private-Key.
Legacy candidates (charge-complementary)
Target Region
Residues 149–153 (0.58 aggregation score)Candidate ID
CP-SOD1-001
(7 residues · computational design)
10/Agent Findings
Literature Agent (1)
These papers are highly relevant to SOD1 G93A-associated ALS as they cover critical aspects including disease mechanisms (protein aggregation, EV signaling, immune dysregulation), therapeutic interventions (tofersen gene therapy, aggregation inhibitors), and genetic interactions that accelerate disease progression. The studies provide both mechanistic insights into how SOD1 mutations drive pathology and evidence for emerging targeted treatments specifically designed for SOD1-ALS patients.
Clinical Agent (1)
No summary available
Structural Agent (1)
AlphaFold structure update: Baseline check: 1 structure(s) found
Supplements Agent (1)
The therapeutic landscape for SOD1 G93A-related ALS shows limited supplement or peptide interventions in active clinical trials, with most current trials focused on gene therapy approaches (ALN-SOD, RAG-17, tofersen). Preclinical research has identified Amisodin as a trimeric SOD1 inhibitor with potential therapeutic effects, though this represents a small molecule drug rather than a traditional supplement. No trials specifically testing dietary supplements, nutritional interventions, or peptide therapeutics for SOD1 G93A were identified in the current landscape.
Synthesis Agent (1)
Synthesis of 5 findings (clinical, literature, peptides, structural, supplements): Recent research on SOD1 G93A-associated ALS reveals a multifaceted therapeutic landscape dominated b...
Peptide Agent (1)
SOD1 G93A: 10 known binders (top: 67.0 nM); 1 candidate peptides designed