01/3D Structure
? About the 3D Viewer
Mol* (pronounced "molstar") is an open-source molecular visualization tool used by the Protein Data Bank and AlphaFold Database. Learn more at molstar.org.
Controls:
- Rotate: Click and drag
- Zoom: Scroll wheel or pinch
- Pan: Right-click and drag (or two-finger drag)
- Reset: Double-click to reset view
What am I looking at?
This is a predicted 3D structure of the protein. The ribbon diagram shows the protein backbone—helices appear as coils, sheets as arrows, and loops as simple lines. The shape determines how the protein functions: where it binds to other molecules, how it catalyzes reactions, and how mutations might disrupt its activity.
Color legend:
The structure is colored by pLDDT confidence score, which indicates how confident AlphaFold is in each region's predicted position:
- Blue (>90): Very high confidence
- Cyan (70-90): Confident
- Yellow (50-70): Low confidence
- Orange (<50): Very low confidence, likely disordered
02/AI Analysis
TLDR
TDP-43 is a protein that normally helps regulate RNA in cells, but when it forms toxic clumps in the brain and spinal cord, it causes ALS (a disease affecting movement) and FTD (a disease affecting behavior and language). This analysis examined the M337V mutation, where methionine at position 337 is replaced by valine, using computer-based structure prediction with moderate confidence (average score 65.7 out of 100). The low confidence scores indicate substantial uncertainty in the predicted structure, limiting definitive conclusions about how this specific mutation alters protein shape and behavior.
Detailed Analysis
Works Cited
Similar Research
03/Research Data
ClinVar Classification
Not found in ClinVar
Population Frequency
No population data available
Disease Associations
2578 totalShowing 5 of 2578 associations
AI Research Brief
Research brief will be generated when agent findings are available.
04/AlphaFold Metrics
05/Domain Annotations
Structural Domains & Regions
Binding Partners
Gene Ontology
06/Structural Caption
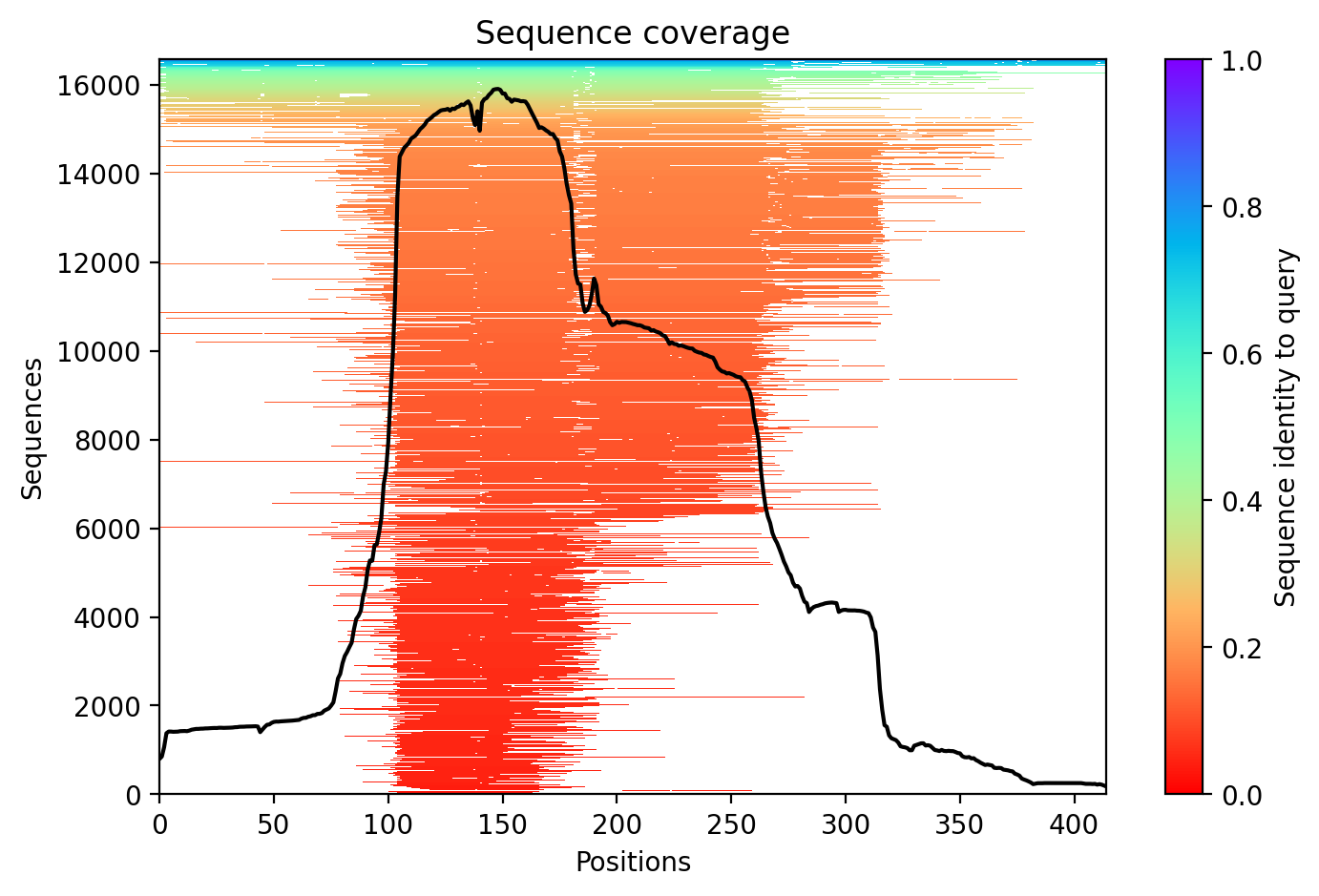
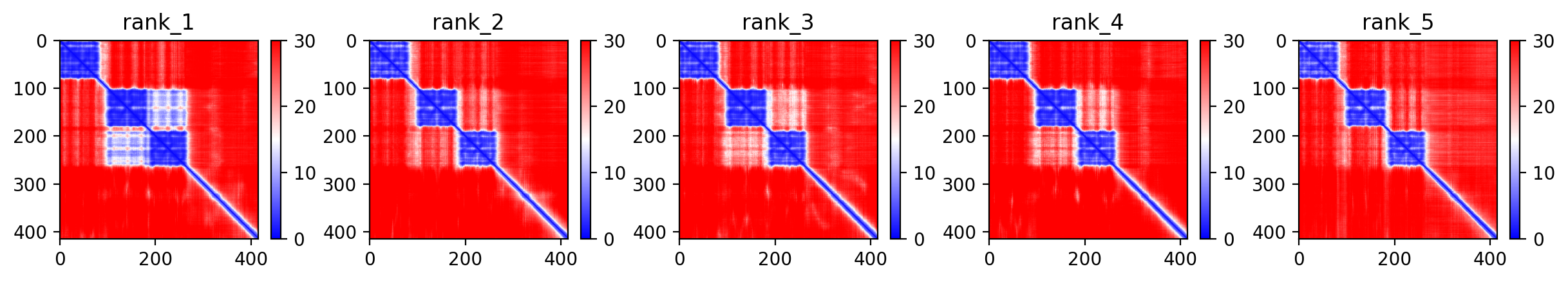
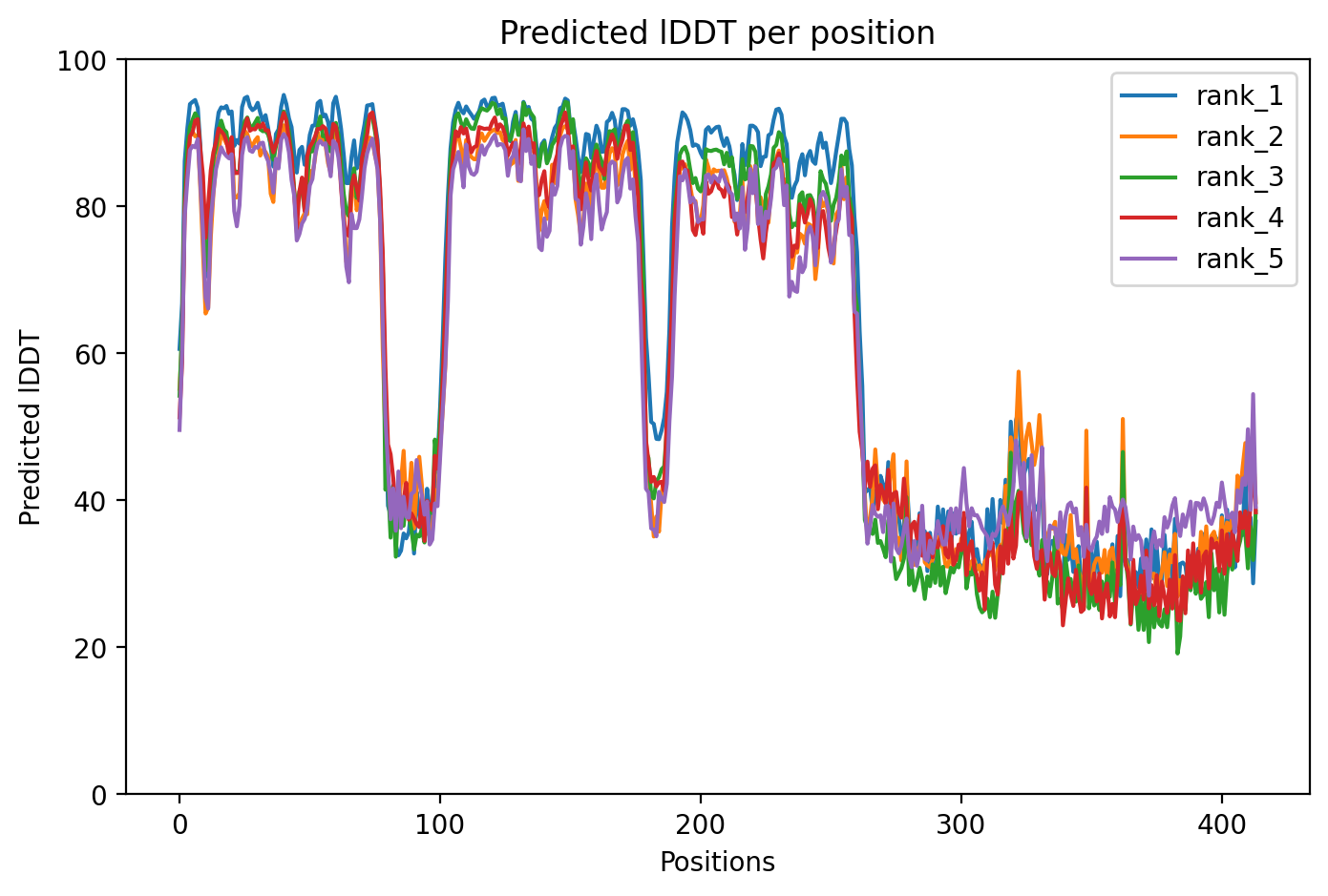
TDP43 M337V structure showing well-folded RRM domains (pLDDT >70) and disordered C-terminal prion-like region where the M337V variant resides (average pLDDT 65.7).
Average pLDDT of 65.7 with 55% high-confidence residues indicates moderate overall structural confidence. Key destabilized regions include the disordered C-terminal domain (residues 261-414) and linker regions between structured RRM domains.
The two RNA recognition motifs (RRM1: 104-200, RRM2: 191-262) show highest confidence and form the structured core. The extensive C-terminal low-complexity region (residues 261-414), including the UBQLN2 interaction domain, glycine-rich tract, and prion-like domain, exhibits low confidence consistent with intrinsic disorder.
The M337V substitution occurs within the intrinsically disordered C-terminal region (residue 337 falls between disordered segments 261-303 and 341-373), likely altering aggregation propensity in the prion-like domain without disrupting the structured RRM cores.
07/Peptide Therapeutics
Aggregation Analysis
Aggregation propensity analysis identifies 1 hotspots (average score: 0.00) using Pawar+KyteDoolittle+charge algorithm.
08/Known Inhibitors
Known Binders from ChEMBL
09/Candidate Peptides
De Novo Peptide Design Pipeline
Pipeline: BoltzGen (de novo binder design) → Boltz-2 rescore → 8-gate wetlab filter → PK + BBB advisory gates. Target site selected from UniProt curated annotations, P2Rank pocket prediction, and aggregation propensity (in that priority order). Advisory gates annotate each candidate with estimated serum half-life, renal/immunogenicity risk, and (for CNS targets) a recommended blood-brain-barrier shuttle conjugation — without silently dropping designs.
Loading candidate statistics...
Sequences are withheld pending IP review. Full candidate data (sequences,
scores, CIF files) is available to authorized reviewers via the
/api/private/candidates/{fold_id} endpoint with
X-Private-Key.
Legacy candidates (charge-complementary)
Target Region
Residues 228–232 (0.71 aggregation score)Candidate ID
CP-TDP43-001
(7 residues · computational design)
10/Agent Findings
Literature Agent (1)
None of the papers directly address the TDP43 M337V variant. The papers focus on other genetic causes of ALS/FTD (C9orf72, CHCHD10, SOD1) and general TDP-43 pathology, but do not mention the M337V mutation in TARDBP. While they provide valuable context about TDP-43 proteinopathy mechanisms and biomarkers relevant to ALS/FTD broadly, they lack specific relevance to understanding the M337V variant's pathogenic effects or clinical characteristics.
Clinical Agent (1)
No summary available
Structural Agent (1)
AlphaFold structure update: Baseline check: 2 structure(s) found
Supplements Agent (1)
The therapeutic landscape for TDP-43 M337V in ALS/FTD shows limited but emerging supplement and peptide interventions. One recruiting trial tests probiotics targeting metabolic pathways in ALS-FTD spectrum disorders. Recent preprints identify TDP-43 aggregation inhibitors and nutritional interventions like caloric restriction that show neuroprotective effects, suggesting potential dietary and peptide-based therapeutic strategies are in early research stages.
Synthesis Agent (1)
Synthesis of 5 findings (clinical, literature, peptides, structural, supplements): The TDP43 M337V variant associated with ALS/FTD presents a mixed therapeutic landscape with emerging...
Peptide Agent (1)
TDP43 M337V: 7 known binders (top: 100.0 nM); 1 candidate peptides designed