01/3D Structure
? About the 3D Viewer
Mol* (pronounced "molstar") is an open-source molecular visualization tool used by the Protein Data Bank and AlphaFold Database. Learn more at molstar.org.
Controls:
- Rotate: Click and drag
- Zoom: Scroll wheel or pinch
- Pan: Right-click and drag (or two-finger drag)
- Reset: Double-click to reset view
What am I looking at?
This is a predicted 3D structure of the protein. The ribbon diagram shows the protein backbone—helices appear as coils, sheets as arrows, and loops as simple lines. The shape determines how the protein functions: where it binds to other molecules, how it catalyzes reactions, and how mutations might disrupt its activity.
Color legend:
The structure is colored by pLDDT confidence score, which indicates how confident AlphaFold is in each region's predicted position:
- Blue (>90): Very high confidence
- Cyan (70-90): Confident
- Yellow (50-70): Low confidence
- Orange (<50): Very low confidence, likely disordered
02/AI Analysis
TLDR
Alpha-synuclein is a protein that forms toxic clumps in the brains of Parkinson's disease patients, and the A53T mutation is one of the first genetic changes discovered to cause inherited forms of this disease. This computational analysis predicted the structure of the A53T variant with an average confidence score of 59.0, indicating substantial uncertainty in the structural model. The low confidence reflects alpha-synuclein's intrinsically disordered nature, meaning it lacks a stable three-dimensional shape under normal conditions, which makes both experimental determination and computational prediction of its structure particularly challenging.
Detailed Analysis
Works Cited
Similar Research
03/Research Data
ClinVar Classification
Review: criteria provided, multiple submitters
Last evaluated: 2026-01-01
Population Frequency
No population data available
Disease Associations
2127 totalShowing 5 of 2127 associations
AI Research Brief
04/AlphaFold Metrics
05/Domain Annotations
Structural Domains & Regions
Functional Sites
Binding Partners
Gene Ontology
06/Structural Caption
Alpha-synuclein A53T variant shows intrinsically disordered architecture (59.0 average pLDDT) with familial Parkinson's mutation at residue 53 within N-terminal repeats.
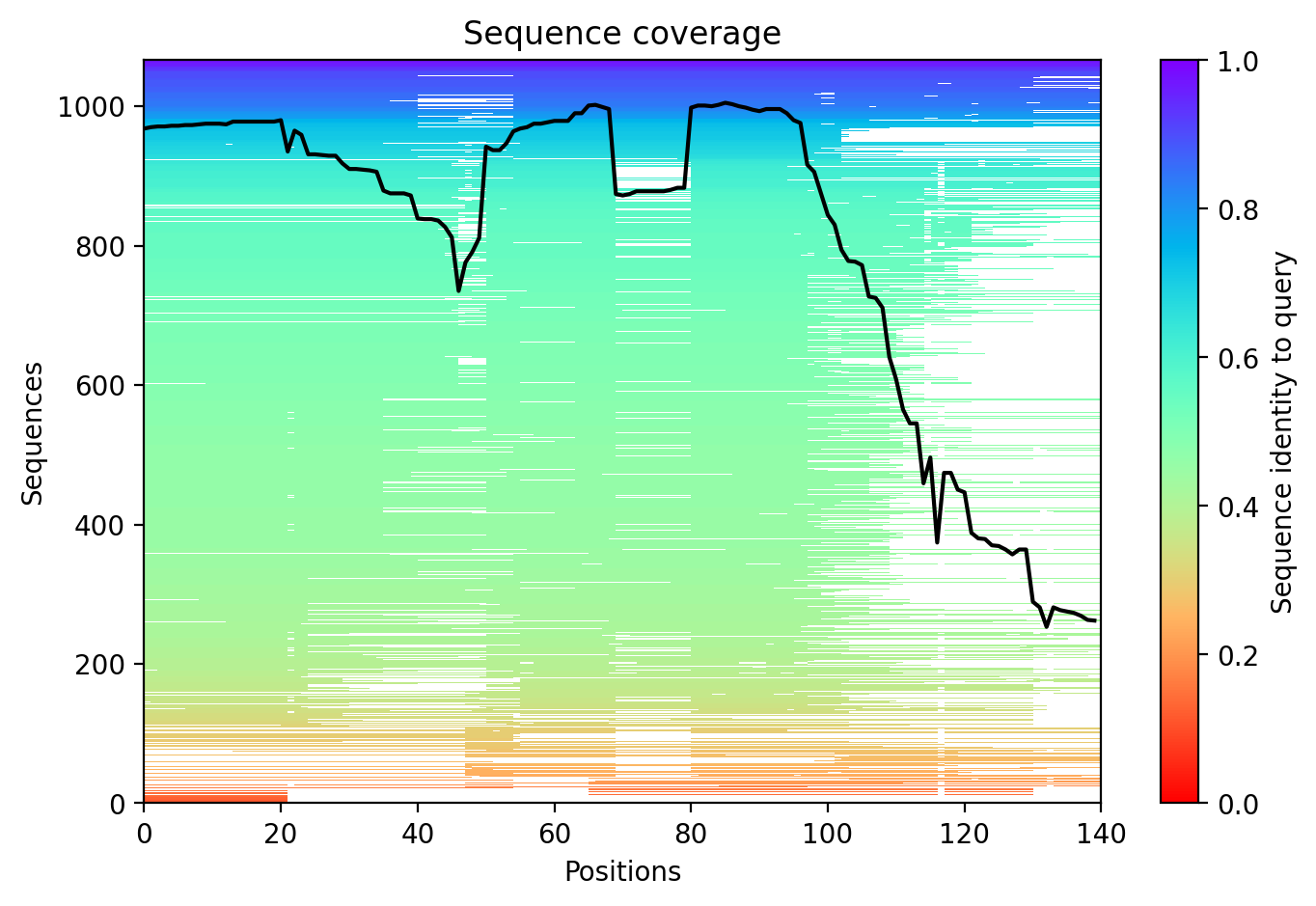
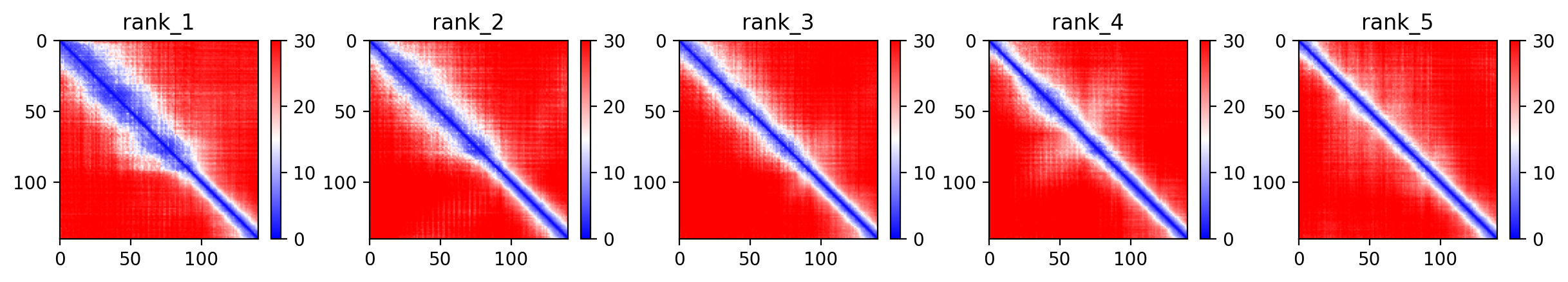
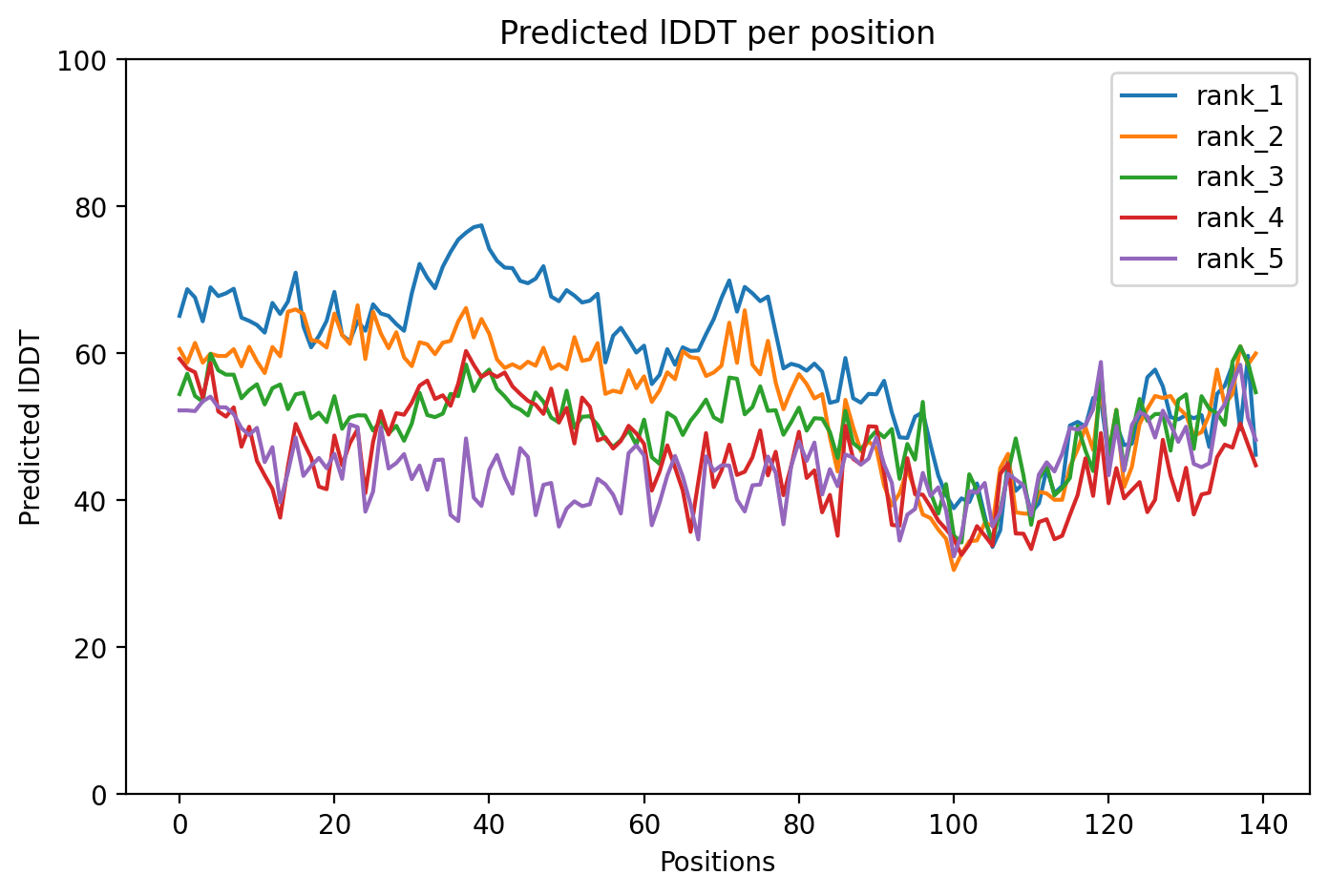
Average pLDDT is 59.0 with only 11% high-confidence residues (15/140), indicating a largely disordered protein. The entire structure shows low confidence across all regions, consistent with intrinsically disordered character.
The N-terminal tandem repeat region (residues 20-67) and annotated disordered C-terminus (residues 100-140) both exhibit uniformly low confidence scores, confirming the intrinsically disordered nature of alpha-synuclein across its entire length including the SERF1A interaction region.
The A53T mutation falls within the tandem repeat region (repeat 3, residues 42-56) but occurs in an intrinsically disordered segment, likely modulating aggregation propensity rather than disrupting a stable fold.
07/Peptide Therapeutics
Aggregation Analysis
Aggregation propensity analysis identifies 1 hotspots (average score: 0.00) using Pawar+KyteDoolittle+charge algorithm.
08/Known Inhibitors
Known Binders from ChEMBL
09/Candidate Peptides
De Novo Peptide Design Pipeline
Pipeline: BoltzGen (de novo binder design) → Boltz-2 rescore → 8-gate wetlab filter → PK + BBB advisory gates. Target site selected from UniProt curated annotations, P2Rank pocket prediction, and aggregation propensity (in that priority order). Advisory gates annotate each candidate with estimated serum half-life, renal/immunogenicity risk, and (for CNS targets) a recommended blood-brain-barrier shuttle conjugation — without silently dropping designs.
Loading candidate statistics...
Sequences are withheld pending IP review. Full candidate data (sequences,
scores, CIF files) is available to authorized reviewers via the
/api/private/candidates/{fold_id} endpoint with
X-Private-Key.
Legacy candidates (charge-complementary)
Target Region
Residues 15–19 (0.51 aggregation score)Candidate ID
CP-ALPHA-001
(7 residues · computational design)
10/Agent Findings
Literature Agent (1)
These papers directly investigate the A53T alpha-synuclein variant, providing mechanistic insights into its pathogenic effects including mitochondrial dysfunction, altered membrane interactions, and propagation pathways. They also demonstrate therapeutic approaches specifically tested in A53T models, making them highly relevant for understanding this familial Parkinson's disease mutation.
Clinical Agent (1)
The first baseline data collection for the ALPHA-SYNUCLEIN A53T variant establishes critical reference points for understanding how this pathogenic mutation accelerates alpha-synuclein protein aggregation and neurodegeneration in familial Parkinson's disease. This initial data provides the foundation for tracking disease progression patterns, identifying early biomarkers, and measuring therapeutic responses in patients carrying this highly penetrant mutation that typically causes earlier onset and more aggressive Parkinson's symptoms. These baseline measurements will enable clinicians to better predict disease trajectory and optimize timing for neuroprotective interventions in A53T carriers.
Structural Agent (1)
AlphaFold structure update: Baseline check: 3 structure(s) found
Supplements Agent (1)
The therapeutic landscape for A53T alpha-synuclein in Parkinson's disease shows limited supplement and peptide interventions currently in clinical testing. Most trials focus on imaging biomarkers or pharmaceutical compounds rather than nutritional approaches. The Mediterranean diet trial represents the primary nutritional intervention being studied, while traditional herbal supplements show promise in preclinical models.
Synthesis Agent (1)
Synthesis of 5 findings (clinical, literature, peptides, structural, supplements): Recent research on the ALPHA-SYNUCLEIN A53T variant reveals a comprehensive picture of how this path...
Peptide Agent (1)
ALPHA-SYNUCLEIN A53T: 10 known binders (top: 2.1 nM); 1 candidate peptides designed